
Современные представления о молекулярных механизмах канцерогенеза при почечно-клеточной карциноме (ПКК) основаны на изучении наследственных форм рака почки. Наследственный, или семейный, рак почки составляет около 4—5 % всех ПКК и относится к проявлениям наследственных онкологических синдромов. Эти синдромы характеризуются герминальными мутациями в соответствующих генах и имеют предрасположенность к определенному гистологическому варианту ПКК [1, 2].
Синдром фон Хиппель—Линдау (VHL) — аутосомно-доминантный синдром, основным проявлением которого служит развитие доброкачественных и злокачественных опухолей в разных органах. По фенотипическим признакам синдром представляется в двух типах. Тип 1 — капиллярные гемангиобластомы сетчатки глаза и ЦНС, а также светлоклеточная ПКК. Тип 2А — капиллярные гемангиобластомы сетчатки глаза, ЦНС и феохромоцитома. Тип 2В — капиллярные гемангиобластомы сетчатки глаза, ЦНС, светлоклеточная ПКК, феохромоцитома и опухоли поджелудочной железы. Тип 2С — только феохромоцитома. При синдроме фон Хиппель—Линдау светлоклеточная ПКК развивается обычно у молодых пациентов и нередко характеризуется мультифокальным ростом и/или билатеральной локализацией. Причиной развития светлоклеточной ПКК при синдроме фон Хиппель—Линдау является герминальная мутация гена-супрессора VHL, расположенного на хромосоме 3р25 [3-4].
Конституциональные транслокации хромосомы 3 — отдельная семейная форма свелоклеточной ПКК, характеризуется сохранением интактной структуры гена VHL и отсутствием «внепочечных» проявлений синдрома фон Хиппель-Линдау [5-6].
Наследственный лейомиоматоз и рак почки - заболевание, характеризующееся развитием лейомиоматоза кожи и матки, папиллярной ПКК 2-го типа и лейомиосаркомой матки. При этом синдроме ПКК обнаруживаются обычно в виде солитарных и унилатеральных узлов, однако отличаются быстрым прогрессированием и ранним метастазированием. Развитие этого заболевания связано с герминальной мутацией гена, локализованного в области 1q42, кодирующего фермент цикла Кребса, фумаратги-дратазу (ФГ). Результаты исследований показывают, что в ФГ-дефицитных ПКК этот фермент выступает в роли опухолевого супрессора и путем двухударной модели Кнадсена участвует в инактивации 2-го аллеля [7-8].
Наследственная папиллярная карцинома почки 1-го типа. Обусловлена герминальными мутациями онкогена МЕТ, локализованного на хромосоме 7q31, что в свою очередь приводит к стимуляции деления клеток и конститутивной активации цитоплазматического домена рецептора. Молекулярная диагностика наследственной папиллярной карциномы почки 1-го типа основывается на обнаружении мутации в экзонах 15-21 гена МЕТ, кодирующих цитоплазматический домен рецептора. При этом заболевании ПКК чаще мультифокальные и билатеральные [9-10].
Синдром Берта-Хогга-Дьюба (BHD) - наследственное аутосомно-доминантное заболевание, которое характеризуется развитием множественных фиброфолликулом кожи лица, шеи, верхней части туловища и кист. Примерно у 35% пациентов с синдромом BHD обнаруживаются разные варианты ПКК, но наиболее часто встречается гибридная онкоцитарная/хромофобная опухоль, или хромофобная ПКК. При этом заболевании опухоли почки, как правило, мультифокальные и билатеральные. При синдроме BHD наблюдается герминальная мутация в гене — супрессоре FLCN, расположенном на хромосоме 17р11.2 [11, 12].
Наследственная параганглиома обусловлена герминальной мутацией в гене субъединицы В сукцинатдегидрогеназы (СДГ). У пациентов с наследственной параганглиомой наблюдаются феохромоцитомы и параганглиомы, а также СДГ-дефицитная ПКК. Изредка встречаются светлоклеточная ПКК, хромофобная ПКК или онкоцитома [13, 14].
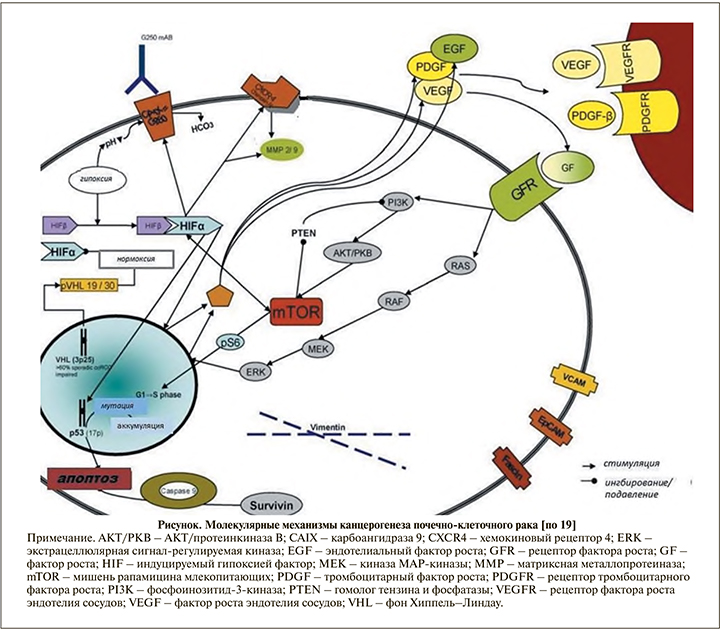
По литературным данным, потеря экспрессии VHL наблюдается в 80% случаев спорадических светлоклеточных ПКК. Как для наследственной формы заболевания, так и для спорадической светлоклеточной ПКК инактивация гена VHL является ключевым событием. Установлено, что в спорадической светлоклеточной ПКК биаллельная инактивация VHL обусловлена точковыми мутациями, аллельными делециями или гиперметилированием промоторной области. Как известно, ген VHL в качестве гена-супрессора контролирует уровень транскрипционных факторов HIF-1a и HIF-2a. В условиях нормальной оксигенации продукт гена VHL (pVHL) в составе E3 убиквитинлигазного комплекса участвует в разрушении транскрипционных факторов путем присоединения убиквитина к гидроксилированным HIF-1a и HIF-2a. В отсутствие pVHL в клетках или в условиях гипоксии продукт гена не связывается с негидроксилированными HIF-1a и HIF-2a, что приводит к накоплению транскрипционных факторов в клетках. При передислокации HIF-a в ядро образуется активный транскрипционный комплекс с HIF-в, вследствие чего запускается механизм активизации генов, связанных с гипоксией: сосудистого эндотелиального фактора роста (VEGF), транспортера глюкозы (GLUT1), тромбоцитарного фактора роста в (PDGFe), рецептора эпидермального фактора роста 1-го типа (EGFR1) и трансформирующего фактора роста (TGFa) [15-18]. В то же время помимо гиперэкспрессии генов, ассоциированных с гипоксией, происходит активация сигнальных путей: сигнального трансдукционного белка (RAS')/гомолога онкогена вирусной мышиной саркомы (Raf)/внеклеточной регулируемой киназы (ERK)/ митогенактивированной протеинкиназы (Мек) и фосфоинозитид-3-киназы (Р/5А)/протеинкиназы В (А_йТ)/мишени для рапамицина млекопитающих (mTOR).
Активация сигнального пути AKT-mTOR является ключевым механизмом в канцерогенезе несветлоклеточных ПКК. При каскадной стимуляции сигнального пути PI3K-AKT-mTOR на фоне связывания факторов роста VEGF и PDGF с их тирозинкиназными рецепторами происходит активация белка 17 mTOR. Внутриклеточная серин/треонинкиназа, именуемая как мишень рапамицина млекопитающих (mTOR), является составной частью двух различных комплексов: TORC1 и TORC2. Комплекс TORC1 стимулирует экспрессию множества белков, регулирующих клеточный цикл (Cyclin D1, c-MYC) и ангиогенез (HIF-1, 2a) путем фосфорилирования ключевых регуляторов трансляции мРНК - 4EBP1 и S6K1-киназы. Комплекс TORC2 поддерживает клеточную полярность и участвует в процессе пространственного роста. Регуляция активности mTOR осуществляется молекулами питательных веществ, различными факторами роста и AKT. Внутриклеточная передача сигнальной кассеты RAS/MAPK является альтернативным путем активации mTOR. Инактивация mTOR наблюдается при нарушении сигнального каскада между PI3K и AKT в результате мутации гена опухолевого супрессора PTEN или вследствие образования комплекса туберозного склероза - TSC1/TSC2. Ряд исследователей отмечают важную роль EGFR-зависимых сигнальных путей в почечном онкогенезе. При этом показана высокая частота экспрессии гена EGFR в различных вариантах ПКК. Схема сигнальных путей, участвующих в онкогенезе ПКК, дана на рисунке [19-22].
Проведенные исследования на цито- и молекулярно-генетических уровнях позволили существенно расширить представление о мутационных процессах при различных гистологических вариантах ПКК.
Светлоклеточная ПКК. При полногеномном секвенировании следующего поколения (next generation sequencing, NGS) 417 образцов светлоклеточной ПКК были выявлены 36 353 соматические мутации, однако по наибольшей частоте повторяемости мутаций было выделено всего 8 генов: VHL, PTEN, BAP1, SETD2, TP53, KDM5C, BRM1 и MTOR. В описанных генах частота мутаций существенно отличалась от таковой в других генах. Приблизительно в 20% светлоклеточной ПКК не были обнаружены мутации ни в одном из этих 8 генов [23, 24].
Мультилокулярная кистозная опухоль почки с низким потенциалом малигнизации по молекулярно-генетическим характеристикам имеет сходство со светлоклеточной ПКК. По литературным данным, примерно в 75% мультилокулярной кистозной опухоли почки с низким потенциалом малигнизации наблюдается потеря короткого плеча хромосомы 3, где расположен ген VHL (регион 3р25). В 25% случаев обнаруживаются соматические мутации или гиперметилирование CрG-островка промоторной области [25-26].
ПКК с мутацией гена TCEB-1 на цитогенетическом уровне характеризуется моносомией по 8-й хромосоме. При NGS 15 образцов ПКК с мутацией гена TCEB-1 были идентифицированы другие мутации в генах Tyr79 (>80%), Ala100, Ala106 и Ser23. При анализе транскриптома показано значительное снижение содержания мРНК генов-POLR2E, POLR2C, CDK7, TCEA1 и TCEB2 [27, 28].
Папиллярная ПКК на цитогенетическом уровне характеризуется увеличением числа копий хромосом 7 и 17, утратой Y-хромосомы. При комплексном генетическом исследовании 161 образца ППКК в 17% опухолей 1-го типа были выявлены точковые мутации гена МЕТ. Среди карцином 2-го типа по наибольшей частоте повторяемости мутаций выделено три гена: SETD2, FH и MiT. В другом исследовании при секвенировании экзома 31 образца в качестве мутаций-драйверов обнаружены изменения в генах ремоделинга хроматина SETD2, BAP1, KDM6A и ARID2. По литературным данным, при папиллярной ПКК мутации в генах SMARCB1 и PBRM1 встречаются с частотой 20-38% [29, 30].
Хромофобные ПКК на цитогенетическом уровне несут потери участков или целых хромосом 1, 2, 6, 10, 13, 17 и 21. При NGS 66 образцов ХПКК было идентифицировано 142 варианта соматических мутаций. Наибольшая частота повторяемости встречалась в генах ND5, P53 и PTEN. Вместе с тем в другом исследовании при полногеномном секвенировании 12 онкоцитом обнаружено, что в них, в отличие от хромофобных ПКК, точковые мутации в генах P53 и PTEN отсутствуют. В связи с этим авторы предположили, что в ходе клональной эволюции инактивирующие мутации в генах P53 и PTEN могут быть причиной трансформации онкоцитомы в эозинофильном варианте ХПКК [31, 32].
Гибридная онкоцитарная/хромофобная опухоль на молекулярно-цитогенетическом уровне характеризуется герминальной мутацией гена FLCN, расположенного на хромосоме 17р11.2. В спорадической форме обнаруживаются моно- и полисомии по хромосомам 1, 2, 6, 9, 10, 13, 17, 21 и 22 [33].
Светлоклеточная папиллярная ПКК. В отличие от светлоклеточной ПКК и папиллярной ПКК, для нее нехарактерны делеция гена VHL и аберрации в хромосомах 7, 17 и Y. При светлоклеточной папиллярной ПКК отмечена гиперэкспрессия мРНК-200 [34, 35].
Тубулокистозная ПКК по молекулярно-генетическому профилю имеет сходство с папиллярной ПКК и характеризуется увеличением числа копий хромосом 7 и 17, утратой Y-хромосомы. При NGS 12 образцов тубулокистозной ПКК мутации были обнаружены в 14 генах. При этом в 60% случаев наибольшая частота повторяемости мутаций была замечена в генах ABL1 и PDFGRA [36-37].
При муцинозной тубулярной и веретеноклеточной ПКК на цитогенетическом уровне обнаруживаются потери участков хромосом 1, 4, 6, 13-15 и 22. В то же время при сравнительной геномной гибридизации определяется увеличение числа копий хромосом 11q, 16q, 17 и 20q. Трисомия по хромосомам 7 и 17 или утрата Y-хромосомы не наблюдаются [38, 39].
ПКК, ассоциированная с приобретенной кистозной болезнью почки на цитогенетическом уровне характеризуется увеличением числа копий хромосом 1, 2, 3, 6, 7, 16, 17 и Y. Наряду с этим, по литературным данным, в этих опухолях в редких случаях обнаруживаются потери участков хромосом 7, 17 или утрата Y-хромосомы [40, 41].
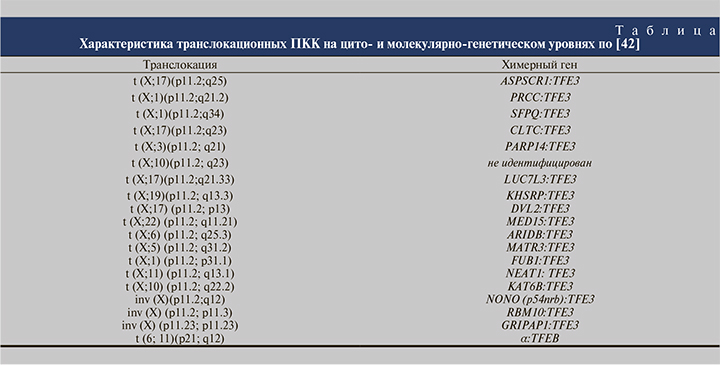
MiTF-ассоциированные ПКК на генетическом уровне обусловлены транслокациями t (X;17)(p11.2;q25), t (X;1) (p11.2;q21), t (X;1)(p11.2;q34), t (X;17)(p11.2;q23), inv (X) (p11.2;q12), t (X;3)(p11.2; q23), t (X;10)(p11.2; q23) и t (6; 11) (p21; q12) с формированием химерных генов (см. таблицу). При комплексном генетическом исследовании в большинстве MiTF-ассоциированных ПКК выявлены соматические мутации в генах MET и BIRC7 [42, 44].
Карцинома из собирательных трубочек на цитогенетическом уровне несут потери участков или целых хромосом 1, 6, 8, 14, 15, 16 и 22. Ряд авторов отмечают в опухоли увеличение числа копий 13q. По литературным данным, амплификация Her-2/neu в карциноме из собирательных трубочек определяется примерно в 45% случаев. Результаты NGS показали, что в этих новообразованиях наблюдаются соматические мутации в генах NF (29%), SETD2 (24%), SMARCB1 (18%) и CDKN2A (12%) [45-46].
Медуллярная карцинома на цитогенетическом уровне характеризуется моносомией 11-й хромосомы. Результаты сравнительной геномной гибридизации показывают, что для медуллярной карциномы характерна утрата гетерозиготности в гене SMARCB1/INI1. Кроме того, в отдельных случаях обнаруживается амплификация генов ABL1 и BCR. В одной из работ в опухоли наблюдалась транслокация t (2;10)(p23;q22) с формированием химерного онкогена VCL-ALK [47-49].
Эозинофильная солидная и кистозная ПКК на генетическом уровне характеризуется транслокациями 16p13.3-16q23.1 (от 33 до 67%), 7p21.2-7q36.2 (от 42 до 50%), 13q14.2 (33%) и 19p12 (3 %), а также делециями Xp11.21 (42%) и 22q11.23 (33%). Потеря гетерозиготности наиболее часто обнаруживалась в хромосомах 16p11.2-11.1 (75%), Xq11.1-13.1 (75%), Xq13.1-21.1 (33%), 11p11.2-11.11 (33%), 9q21.1-22.2 (33%) и 9q33.1 (33%). В одной из работ при исследовании 7 образцов спорадической эозинофильной солидной и кистозной ПКК методом NGS в 5 и 2 случаях наблюдались соматические мутации в генах TSC2 и TSC1 соответственно [50, 51].
Фолликулярная карцинома почки, СДГ-дефицитная ПКК и ФГ-дефицитная ПКК, по данным литературы, на цитогенетическом уровне не имеют характерных хромосомных аберраций. Между тем в одном исследовании при сравнительной геномной гибридизации одного образца фолликулярной карциномы обнаружено увеличение числа копий хромосом 7q36, 8q24, 12, 16, 17p11-q11, 17q24, 19q, 20q13, 21q22.3 и утрата 1p36.3, 9q21-33 и Х-хромосомы [52-54].
В настоящее время активно обсуждается роль раковых стволовых клеток (РСК) в молекулярном патогенезе ПКК. РСК - небольшая устойчивая субпопуляция клеток, способных к постоянному самообновлению и дифференцировке. Такие стволовые клетки, также известные как опухольинициирующие клетки, обладают высокой степенью резистентности и часто выживают после химио- и лучевой терапии. Изучение свойств РСК и механизмов их регуляции открывает большие перспективы в разработке новых подходов к противоопухолевой терапии. По данным ряда исследователей, среди потенциальных диагностических маркеров РСК ПКК большой интерес представляют ALDH1A1, CXCR4, CD24, CD82, CD105, CD133, SOX-2, NANOG и OCT4 [55-57].
Таким образом, ПКК представляют собой широкую и гетерогенную группу заболеваний. С каждым последующим пересмотром классификация этих опухолей неуклонно расширяется, появляются все новые критерии дифференциальной диагностики с применением молекулярных маркеров. Внедрение в морфологическую практику усовершенствованных алгоритмов молекулярно-генетической диагностики сможет обеспечить высокую диагностическую достоверность в определении конкретной нозологической формы ПКК, а в конечном итоге более достоверно прогнозировать течение заболевания.


