
Мочекаменная болезнь (МКБ) – хорошо известное и широко распространенное заболевание мочевыделительной системы с высокой частотой рецидивирования [1, 2]. В настоящее время не вызывает сомнений весомый вклад генетических факторов в развитие МКБ.
Как правило, МКБ представлена полигенными формами уролитиаза. Диагностика этих форм представляет определенные трудности вследствие взаимодействия множества средовых и генетических факторов, приводящих к формированию МКБ. Другие случаи МКБ представлены моногенными формами заболевания, которых на сегодняшний день описано более 80 [3]. Моногенные формы МКБ можно диагностировать более точно ввиду наличия конкретного дефекта в определенном гене [4].
Большинство моногенных форм уролитиаза обусловлено нарушением кальциевофосфорного обмена, что подтверждается более частым выявлением камней, в химический состав которых входит кальций (85% – оксалат кальция, до 10% – фосфат кальция, 5% – оксалат и фосфат кальция в сочетании с мочевой кислотой). Более редкие – мочекислые (5–10%), струвитные (5–15%) и цистиновые (1–3%) камни [5, 6].
Моногенные формы уролитиаза могут проявляться в разном возрасте, однако чаще они манифестируют в раннем детстве. В большинстве случаев ведущим симптомом заболевания является камнеобразование в почках, но некоторые нозологические формы могут сопровождаться нарушением работы других органов и систем [3].
Применение современных молекулярно-генетических технологий позволяет проводить раннюю диагностику, своевременное лечение и профилактику заболевания в семье больного.
В качестве молекулярно-генетических технологий используют полимеразную цепную реакцию (ПЦР), секвенирование по Сэнгеру, секвенирование нового поколения (NGS, от англ. Next Generation Sequencing) в виде таргетных NGS-панелей, клинического экзома, полноэкзомного секвенирования, полногеномного секвенирования.
В настоящей работе представлены описания клинических наблюдений моногенных форм уролитиаза, в диагностике которых использованы разные виды анализа ДНК.
Под нашим наблюдением находились 5 пациентов с диагнозом «медуллярный нефрокальциноз», или МКБ, установленным в детском возрасте. Возраст пациентов от 1 года до 9 лет. Всем пациентам проведено инструментальное обследование (УЗИ органов брюшной полости, почек, мочевого пузыря), биохимическое исследование крови (показатели кислотно-щелочного состояния, минерального обмена и электролитного состава: кальций, фосфор, магний, натрий, калий, хлориды, железо), паратиреоидный гормон (ПТГ), 25(ОН)D) и мочи (показатели кристаллурии с обязательной оценкой кальциево-креатининового соотношения), а также выполнен спектральный анализ камней.
Для выявления генетических причин МКБ в зависимости от клинической картины заболевания больным проведено ДНК-исследование с использованием полноэкзомного секвенирования и таргетных NGS-панелей двух видов: 1) гены CYP24A1, SLC34A1; 2) гены FGF23, PTHR1, CASR, SLC34A1, ATP6V1B1, SLC9A3R1, SLC34A3, CYP24A1, ATP6V0A4, GALNT3, ALPL, PHEX, CYP27B1, LRP5, DMP1, VDR, KL, CYP2R1, ENPP1, CLCN5, CLCNKB, SLC2A2 (с общим покрытием 98,5%).
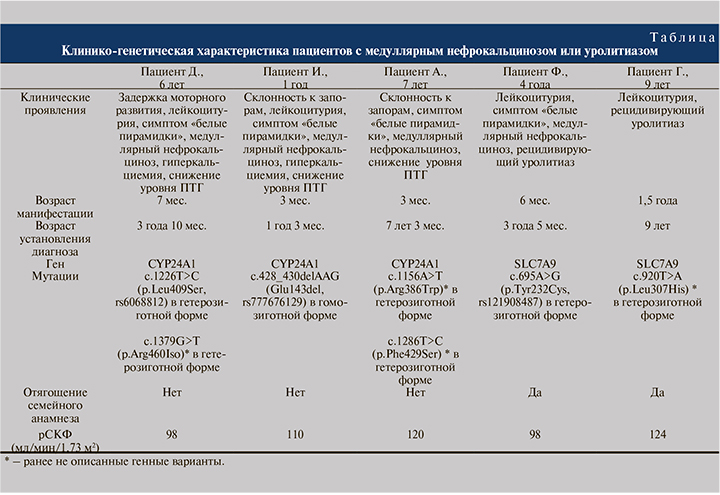
Пациент Д., мальчик 6 лет. При рождении масса тела составляла 3350 г, рост – 51 см. На первом году жизни отмечены задержка моторного развития, гипотония; наблюдался у невролога, по результатам инструментальных методов исследования (электронейромиография) данных за нервно-мышечные заболевания не получено.
К нефрологу впервые обратились в возрасте ребенка 7 мес. в связи с постоянной лейкоцитурией в анализах мочи. При УЗИ почек выявлен синдром белых пирамидок. При биохимическом исследовании крови выявлена гиперкальциемия – Саобщ максимально до 3,36 ммоль/л (норма – 2,2–2,7 ммоль/л) и Са++ – 1,7 ммоль/л (норма – 1,13–1,32 ммоль/л), снижение уровня ПТГ до 3 пг/мл (норма – 16–62 пг/мл), фосфор – 1,57 ммоль/л (норма – 1,3–2,26 ммоль/л), креатинин – 40 мкмоль/л, рСКФ (по формуле Шварца – 98 мл/мин/1,73 м2. При биохимическом исследовании мочи получены следующие данные: Ca/Cr – 0,8 ммоль/ммоль (норма до 1,1 ммоль/ммоль), мочевая кислота/Cr – 0,65 ммоль/ммоль (норма до 1,0 ммоль/ммоль), оксалаты/Cr – 50 мкмоль/ммоль (норма до 82 мкмоль/ммоль), белок – 0,3 г/сут. Уровень 25(ОН)D составил 21,1 нг/мл (норма – 14–60 нг/мл). Клинический диагноз: медуллярный нефрокальциноз, цистит, гипопаратиреоз.
В возрасте 3 лет 10 мес. пациенту проведено молекулярно-генетическое исследование методом массового параллельного секвенирования по панели генов, включившей 22 гена, связанных с нарушением кальциевофосфорного обмена. В результате секвенирования в гене CYP24A1 выявлено два варианта: известная патогенная мутация c.1226T>C (p.Leu409Ser, rs6068812) и ранее неописанный вариант c.1379G>T (p.Arg460Iso), оба в гетерозиготной форме. Обнаруженные находки соответствуют идиопатической инфантильной гиперкальциемии 1-го типа.
Пациент И., мальчик 1 года. Масса тела при рождении – 3300 г, рост – 52 см. С 1-го месяца отмечена склонность к запорам, с 3 мес. – снижение аппетита, плоская весовая кривая, периодическая рвота. С 5 мес. регистрировали лейкоцитурию до 50 в поле зрения. В то же время при УЗИ почек выявлены признаки медуллярного нефрокальциноза в виде симптома «белые пирамидки». В биохимическом анализе крови выявлена гиперкальциемия (Саобщ максимально до 3,08 ммоль/л (норма – 2,20–2,70 ммоль/л), фосфор – 1,75 ммоль/л (норма – 1,3–2,26 ммоль/л), ПТГ менее 3 пг/мл (норма – 16–62 пг/мл), креатинин – 24,3 мкмоль/л, рСКФ – 110 мл/мин/1,73 м2. При биохимическом исследовании суточной мочи: Ca/Cr – 0,67 мг/мг (норма до 0,8 мг/мг), мочевая кислота/Cr – 0,9 мг/мг (норма до 2,2 мг/мг), оксалаты/Cr – 90 мг/г (норма до 140), фосфор/Cr – 1,8 мг/мг (норма до 4 мг/мг).
В ходе осмотра неврологом выявлена диффузная мышечная гипотония. Клинический диагноз: медуллярный нефрокальциноз, инфекция органов мочевой системы, гипопаратиреоз. Синдром вялого ребенка (диффузная мышечная гипотония). С целью выявления генетической причины заболевания и уточнения диагноза в возрасте 1 года 3 мес. пациенту проведено ДНК-исследование по панели генов, включившей 22 гена, связанных с нарушением кальциевофосфорного обмена. В результате секвенирования в гене CYP24A1 выявлена известная патогенная мутация c.428_430delAAG (Glu143del, rs777676129) в гомозиготной форме. Установленный генотип подтверждает диагноз идиопатической инфантильной гиперкальциемии 1-го типа.
Пациентка А., девочка 7 лет. Масса тела при рождении – 3000 г, рост – 50 см. Рождена от первой беременности, протекавшей с угрозой прерывания на ранних сроках.
В 6 лет обратилась с жалобами на боли в животе. В возрасте 1 мес. у ребенка отмечена склонность к запорам, с 3 мес. появилась периодическая рвота, с 5 мес. выявлена лейкоцитурия до 50 в поле зрения. В то же время по данным УЗИ почек выявлены признаки медуллярного нефрокальциноза в виде симптома «белые пирамидки». Проведена симптоматическая терапия с положительным эффектом. В возрасте 6 лет проведено обследование. В анализах крови: Саобщ максимально до 2,42 ммоль/л (норма – 2,20–2,70 ммоль/л), фосфор – 1,59 ммоль/л (норма – 1,3–2,26 ммоль/л), ПТГ менее 3 пг/мл (норма – 16–62 пг/мл), 25(ОН)D – 55,5 нг/мл (норма – 14–60 нг/мл), креатинин – 49,6 мкмоль/л, рСКФ – 120 мл/мин/1,73 м2.
Биохимическое исследование суточной мочи: Ca/Cr – 0,27 мг/мг (норма до 0,21 мг/мг), мочевая кислота/Cr – 0,74 мг/мг (норма до 0,9 мг/мг), оксалаты/Cr –24 мг/г (норма до 60 мг/г), фосфор/Cr – 1,35 мг/мг (норма до 0,97 мг/мг).
Клинический диагноз: медуллярный нефрокальциноз, гипопаратиреоз, гиперкальциурия. В возрасте 7 лет 3 мес. проведен молекулярно-генетический анализ по панели генов, включившей 22 гена, связанных с нарушением кальциево-фосфорного обмена, который выявил в гене CYP24A1 наличие двух ранее не описанных генных вариантов: c.1156A>T (p.Arg386Trp) и c.1286T>C (p.Phe429Ser). Обнаруженные молекулярно-генетические находки могут быть причиной идиопатической инфантильной гиперкальциемии 1-го типа.
Идиопатическая инфантильная гиперкальциемия 1-го типа – аутосомно-рецессивное наследственное заболевание, вызванное мутациями гена CYP24A1. Ген CYP24A1 расположен на длинном плече хромосомы 20 (20q13.2) и кодирует фермент внутренней мембраны митохондрий 24-гидроксилазу, активация которого приводит к разрушению 25-гидроксихолекальциферла (25-(OH)D3) и кальцитриола (1,25-(OH)2D3). При нарушении функции данного гена наблюдаются высокий уровень активности витамина D и как следствие – увеличение всасывания кальция в кишечнике, гиперкальцеимия и гиперкальциурия. Идиопатическая гиперкальциемия в большинстве случаев манифестирует в возрасте 4–12 мес., но может проявляться с рождения. Кроме перечисленных симптомов у пациентов могут наблюдаться задержка развития, рвота, обезвоживание, медуллярный нефрокальциноз [7, 8]. Лечение при идиопатической инфантильной гиперкальциемии симптоматическое. Родители должны строго следить за соблюдением питьевого режима (около 30–50 мл/кг/сут дробно в течение дня), ограничивать использование продуктов, содержащих большое количество витамина D, а также комплексных витаминных препаратов, избегать избыточной инсоляции и в обязательном порядке использовать защитные средства для кожи с высоким SPF-фактором. Употреблять кальцийсодержащие продукты можно, но в количествах, не превышающих возрастные нормы [9, 10]. Назначение кетоконазола (как ингибитора цитохрома Р-450, который также ингибирует 25-гидроксилазу и 1-альфа-гидроксилазу), гемодиализа при угрожающей гиперкальциемии, бисфосфонатов используется крайне редко и описано в качестве подхода к лечению таких пациентов лишь в единичных публикациях [11].
В терапии также могут быть использованы цитратные препараты в качестве ингибиторов кристаллообразования и средств снижения выраженности проявлений нефрокальциноза [12].
Пациент Ф., мальчик 4 лет, родился от I беременности с массой тела 4110 г, ростом 56 см. Роды с акушерским пособием (метод Кристеллера). С 6 мес. отмечались гипертермия, в анализах – лейкоцитурия. При УЗИ почек с 6 мес. выявлялся симптом «белые пирамидки». В анализах крови показатели кальция (общего и ионизированного), фосфора в пределах нормальных значений. В 3 года на фоне болевого синдрома произошло самостоятельное отхождение почечного камня. Рентгенофазовый анализ уролита выявил оксалат кальция моногидрат (вевеллит) – 23%, оксалат кальция гидрат (ведделит) – 28%, дигидроксофосфат кальция (гидроксиапатит) – 32%, фосфат кальция (витлокит) – 17%. Семейный анамнез отягощен: МКБ у матери с 27 лет и у бабки по материнской линии с 40 лет. Биохимический анализ крови: Саобщ – максимально 2,66 ммоль/л (норма – 2,20–2,70 ммоль/л), Са++ – 1,26 ммоль/л, фосфор – 1,63 ммоль/л (норма –1,3–2,26 ммоль/л), ПТГ – 13,5 пг/мл (норма – 16–62 пг/мл), 25(ОН)D – 56,8 нг/мл (норма – 14–60 нг/мл), креатинин – 47 мкмоль/л, рСКФ – 98 мл/мин/1,73 м2. Клинический диагноз: медуллярный нефрокальциноз, пиелонефрит, гипопаратиреоз. В возрасте 3 лет 5 мес. пациенту проведено секвенирование экзома, по результатам которого в гене SLC7A9 выявлена мутация c.695A>G (p.Tyr232Cys, rs121908487) в гетерозиготной форме.
Мутации гена SLC7A9 ответственны за развитие цистинурии – моногенного заболевания, которое в зависимости от типа мутации может наследоваться аутосомно-рецессивно, аутосомно-доминантно, а также возможен дигенный тип наследования. Выявленная мутация в гене SLC7A9 на сегодняшний день классифицируется как type non-I, т.е. в гетерозиготной форме обычно не приводит к клиническому проявлению заболевания в виде образования цистиновых камней [13]. Однако есть данные о том, что в гетерозиготной форме мутации в гене SLC7A9 могут способствовать камнеобразованию у пациентов, в том числе могут образовываться камни, в составе которых преобладают кальциевые соли. Последнее связано с тем, что цистин мочи сам по себе способствует кристаллизации кальция в мочевыводящих путях [14]. Таким образом, полученные данные полноэкзомного секвенирования пациента соответствовали клинической картине данной формы МКБ.
Пациент Г., девочка 9 лет от первой беременности, протекавшей на фоне анемии, диффузно-токсического зоба 2-й ст. Роды в срок, масса тела при рождении – 3380 г, рост –51 см. С возраста 1,5 лет в анализах мочи отмечались лейкоцитурия до 10–15 в поле зрения, кристаллы цистина, ураты, оксалаты. При УЗИ почек выявлены правосторонняя пиелоэктазия и конкременты до 3 мм. В возрасте 2 лет ребенку в связи с правосторонним уролитиазом проведена нефролитотрипсия. При контрольном УЗИ почек через 12 мес. определялись правосторонняя пиелоэктазия до 23 мм, множественные конкременты 6–18 мм; по результатам КТ брюшной полости в правой лоханке выявлен конкремент размером 20×13×19 мм, в чашках визуализировались множественные конкременты размером от 5 до 20 мм. Биохимический анализ крови: Саобщ общий – максимально 2,48 ммоль/л (норма – 2,20–2,70 ммоль/л), Са++ – 1,26 ммоль/л (норма – 1,13–1,32 ммоль/л), фосфор – 1,35 ммоль/л (норма – 1,3–2,26 ммоль/л), ПТГ – 94,5 пг/мл (норма – 16–62 пг/мл), 25(ОН)D – 14 нг/мл (норма – 14–60 нг/мл), креатинин – 42 мкмоль/л, рСКФ – 124 мл/мин/1,73 м2. С 2012 по 2017 г. в связи с рецидивирующим уролитиазом ребенку трижды проведена дистанционная литотрипсия. Данные тонкослойной хромотографии мочи соответствовали умеренной неселективной гипераминоацидурии. Рентгенофазовый анализ уролита: 100%-ный вевелит. Семейный анамнез отягощен по материнской линии: МКБ у деда и у троюродного дяди по материнской линии. В возрасте 9 лет ребенку проведено молекулярно-генетическое исследование в виде секвенирования экзома, по результатам которого выявлен ранее не описанный вариант c.920T>A (p.Leu307His) в гене SLC7A9 в гетерозиготной форме, который может быть причиной цистинурии.
Цистинурия – аутосомно-рецессивное заболевание, которое встречается в 1–10% случаев МКБ у детей [15–17]. Распространенность заболевания в мире составляет 1:7000 [18]. Цистинурия характеризуется гиперэкскрецией цистина с мочой (более 400 мг в день при норме менее 30 мг в день), рецидивирующим течением МКБ и образованием в почках цистиновых уролитов [19]. Клинически заболевание манифестирует в возрасте от 2 до 40 лет. В 75% случаев наблюдается двустороннее поражение почек [20]. Цистинурия возникает из-за дефекта в системе трансэпителиальных транспортеров двуосновных аминокислот (цистина, орнитина, лизина и аргинина) в проксимальных почечных канальцах и в желудочно-кишечном тракте. Цистин плохо растворим в обычной моче с pH менее 7. В результате перенасыщения мочи цистин подвергается кристаллизации с образованием камней, чаще всего имеющих цистиновый состав, в некоторых случаях уролиты представлены солями кальция [21, 22].
Лечение цистинурии предусматривает повышение растворимости цистина посредством увеличения pH и объема мочи. Увеличение диуреза (полиурия) служит одним из наиболее эффективных способов предотвращения развития почечных камней у таких пациентов. Тиоловые производные D-пеницилламин и тиопронин используют в наиболее тяжелых случаях. Эти лекарственные средства содержат сульфгидрильные группы и образуют хорошо растворимые соединения с цистином (цистеин-пеницилламин и цистеин-тиопронин) [23, 24].
Как видно из представленных клинических наблюдений (см. таблицу), МКБ может манифестировать в раннем детском возрасте и на начальном этапе проявляться ультразвуковым симптомом «белые пирамидки», который является отражением медуллярного нефрокальциноза и свидетельствует о высоком риске формирования камней в почках [25].
Чаще всего дети поступают под наблюдение нефролога в связи со случайным обнаружением при УЗИ почек симптома «белые (гиперэхогенные) пирамидки» – медуллярного нефрокальциноза, который может быть признаком ряда состояний (медуллярная кистозная болезнь, болезнь Дента, дистальный почечно-тубулярный ацидоз [ПТА], семейная гипомагнезиемическая гиперкальциурия с нефрокальцинозом, синдром Вильямса, синдром Беквитта–Видемана и др.). Кроме того, это состояние может быть обусловлено ятрогенными причинами (передозировка витамина D, молочно-щелочной синдром и др.) и проявлениями гиперкальциурии при онкологических заболеваниях [12, 26].
На первом этапе обычно проводят оценку кислотно-основного состояния крови, биохимическое исследование крови (креатинин, мочевина, щелочная фосфатаза, кальций общий и ионизированный, фосфор, ПТГ-гормон, 25(ОН)D (и по возможности его метаболит 1,25(ОН)2D, магний, натрий, хлор, калий), а также биохимическое исследование мочи (креатинин, кальций, оксалаты, мочевая кислота, фосфор, общий белок).
При наличии гиперкальциурии, гиперкальциемии, гипопаратиреоза, нормального уровня 25(ОН)D и повышенного уровня 1,25(ОН)2D можно заподозрить идиопатическую инфантильную гиперкальциемию. Характерными признаками других болезней, сопровождающихся уролитиазом, служат низкомолекулярная протеинурия при болезни Дента; метаболический ацидоз при дистальном почечно-тубулярном ацидозе (ПТА); низкий уровень магния и азотемия при гипомагнезиемической гиперкальциурии с нефрокальцинозом; гипокалиемический гиперхлоремический алкалоз при синдроме Барттера (неонатальная форма сопровождается гиперкальциурией и развитием нефрокальциноза); низкий уровень щелочной фосфатазы при гипофосфатазии; специфические изменения внутренних органов и внешности при синдромальных формах и цилиопатиях [12].
Однако клиническая и лабораторно-инструментальная диагностика не всегда позволяет установить точную причину заболевания. В связи с этим в настоящее время в диагностике МКБ широко используются молекулярно-генетические технологии. Причем в каждом случае заболевания выбор молекулярно-генетического метода должен проводиться индивидуально.
Так, если заболевание связано с известными частыми мутациями в определенном гене, то назначается ДНК-диагностика на основе полимеразной цепной реакции (ПЦР). В случае редких заболеваний, для генов которых не описаны повторяющиеся мутации, используются методы секвенирования ДНК, представленные севенированием по Сэнгеру и NGS. Вследствие широкого клинического полиморфизма и генетической гетерогенности причин моногенных форм уролитиаза в ряде случаев целесообразно исследование не одного, а сразу нескольких генов.
В таких случаях используют таргетные NGS-панели генов [3]. В отсутствие патогенных мутаций в исследуемых генах таргетной панели применяют более широкое генетическое обследование в виде полноэкзомного или полногеномного секвенирования. Применение молекулярно-генетических методов служит важным дополнением в диагностике моногенных форм уролитиаза.
Современный уровень развития медицины позволяет успешно применять разнообразные молекулярно-генетические методы в диагностике мочекаменной болезни. Особое значение молекулярно-генетические технологии имеют при ранней манифестации заболевания, а также при отягощенном по МКБ семейном анамнезе. Выявление точной молекулярной причины МКБ дает возможность проводить эффективную профилактику в семье больного, а также в перспективе обеспечить индивидуальный подход к терапии. В связи с этим не вызывает сомнений необходимость медико-генетического консультирования пациентов с МКБ.


