
Расшифровка структуры генома человека открыла путь к пониманию молекулярных основ болезней, разработке принципиально новых стратегических подходов к их диагностике и лечению. Не стала исключением и мочекаменная болезнь (МКБ).
Мочекаменная болезнь занимает одно из ведущих мест в структуре урологических заболеваний и представляет одну из актуальных проблем современной урологии в связи с высокой распространенностью и склонностью к рецидивированию [1, 2]. Уролитиаз является третьим по встречаемости урологическим заболеванием, частота которого составляет 15–25%. Распространенность нефролитиаза среди населения России составляет у детей 19–20 случаев, у подростков – 80–82, у взрослых – 450–460 случаев на 100 тыс. населения. Примерно в 65–70% случаев болезнь диагностируют у лиц в возрасте 20–25 лет, т.е. в наиболее трудоспособном периоде жизни [3, 4].
В подавляющем большинстве случаев, практически в 80%, конкременты имеют в своем химическом составе кальций (85–90% – оксалатно-кальциевые, 1–10% – фосфатно-кальциевые, 5% – оксалат и фосфат кальция в сочетании с мочевой кислотой). На мочекислые камни приходится 5–10%, на струвитные – 5–15%, на цистиновые – 1–3% [5, 6]. Несмотря на внедрение современных методов лечения (дистанционная литотрипсия, контактная уретеролитотрипсия, чрескожная нефролитотрипсия), частота рецидивов остается на довольно высоком уровне и достигает 38,4–50% [7, 8]. В связи с ростом распространенности заболевания остается актуальным поиск факторов риска камнеобразования и изучение роли наследственности и генетических критериев диагностики. К настоящему времени убедительно доказана тесная взаимосвязь генетических нарушений с клиническими проявлениями практически всех заболеваний человека, в том числе МКБ. При этом уролитиаз может иметь моногенную природу наследования, а может быть связан с генетической предрасположенностью, в основе которой лежит действие множества генетических факторов во взаимодействии их между собой и с факторами экзогенной природы.
В настоящей работе будут рассмотрены моногенные формы заболевания.
По данным каталога В. А. Маккьюсика (www.omim.org), существует по меньшей мере 80 моногенных форм уролитиаза. В основном они относятся к редким наследственным заболеваниям, однако в силу концентрации таких больных на приеме многие из них хорошо известны практикующим врачам-урологам (болезнь Дента, синдром Барттера, почечный канальциевый ацидоз и др.).
Стоит отметить, что гиперкальциурия и гипероксалурия считаются наиболее важными факторами риска МКБ. Из всех нарушений состава мочи чаще всего при МКБ встречается гиперкальциурия, которая наблюдается у 40–50% больных. Ниже представлена информация о генетической составляющей развития уролитиаза, связанного именно с нарушением кальциевого обмена.
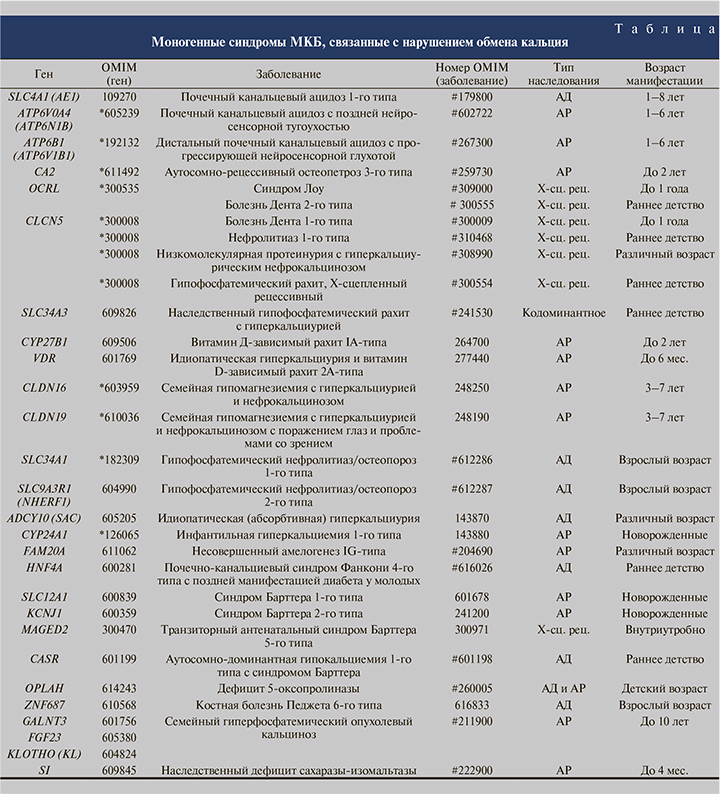
Моногенные формы МКБ, сопровождающиеся образованием кальциевых камней, в основном являются редкими заболеваниями и чаще проявляются сразу после рождения или в первые годы жизни ребенка. Однако среди них немало форм, имеющих различный возраст манифестации. В большинстве случаев ведущим симптомом заболевания выступает непосредственно камнеобразование в почках, тогда как некоторые нозологические формы сопровождаются дополнительными изменениями в других органах и системах. Не вызывает сомнений важность ранней и точной диагностики патологии с использованием молекулярно-генетических методов в связи с необходимостью своевременного назначения соответствующей терапии и профилактики заболевания у родственников пациента. В настоящее время молекулярно-генетическая диагностика уже успешно применяется для верифицирования наследственных синдромов, связанных с образованием кальциевых камней, которые встречаются в клинической практике врача-уролога. В таблице представлены примеры моногенных синдромов МКБ, связанных с преобладанием кальциевых камней.
Как видно из таблицы, для ряда моногенных форм МКБ характерно явление генетической гетерогенности молекулярных причин заболевания. Так, например, аутосомно-рецессивный семейный туморальный гиперфосфатемический кальциноз может быть связан с мутациями в различных генах, в частности в генах GALNT3, FGF23, KL. При этом симптоматика и возраст дебюта заболевания будут однотипными во всех представленных случаях.
В то же время различные мутации в одном гене могут приводить к развитию разнообразных симптомокомплексов. Например, мутации гена CLCN5, экспрессирующегося преимущественно в почках и кодирующего хлорный канал 5-го типа, приводят к развитию разных заболеваний, а именно болезни Дента 1-го типа, низкомолекулярной протеинурии с гиперкальциурическим нефрокальцинозом и гипофосфатемического рахита. Все патологии наследуются Х-сцепленнорецессивно, но имеют разный возраст манифестации и различные клинические проявления (см. таблицу).
Ниже приведено описание наиболее часто встречающихся в практике врачей-урологов моногенных форм кальциевого уролитиаза.
Болезнь Дента – заболевание, характеризующееся Х-сцепленным рецессивным типом наследования. Выделяют две формы патологии: болезнь Дента 1-го и 2-го типов. Лабораторные показатели пациентов с обоими типами патологии схожи. В целом для заболевания характерно наличие низкомолекулярной протеинурии (один из патогномоничных признаков болезни Дента), гиперкальциурии, гипофосфатемии, гиперфосфатурии, аминоацидурии, увеличение уровня витамина D3 в крови, может выявляться микрогематурия. Клинически у пациентов наблюдается низкий рост (чаще при болезни Дента 1-го типа), постепенно развиваются нефрокальциноз, нефролитиаз и почечная недостаточность. Обе формы характеризуются ранним началом, однако в случае болезни Дента 2-го типа у некоторых детей дополнительно может наблюдаться неврологическая симптоматика, в частности нарушение умственного развития. Почечные депозиты состоят из кальция фосфата и кальция оксалата. Первый тип болезни Дента обусловлен мутациями в упомянутом выше гене хлоридного канала 5-го типа (CLCN5), в то время как болезнь Дента 2-го типа связана с мутациями другого гена, OCRL, кодирующего фосфатидилинозитол-4,5-бисфосфат-5-фосфатазу и вовлеченного в полимеризацию актина. В перечисленных генах описаны в основном индивидуальные семейные мутации [9–11]. Исходя из Х-сцепленного рецессивного типа наследования патологии, в полной мере заболевание проявляет себя только у лиц мужского пола. У 30–80% пациентов (чаще мужского пола) почечная недостаточность достигает своей терминальной стадии к возрасту 30–50 лет. Вместе с тем известно, что у женщин с мутациями гена CLCN5 в гетерозиготной форме также могут выявляться некоторые изменения состава крови и мочи: асимптомная протеинурия, гиперкальциурия или изолированная гипофосфатемия, которые часто не сопровождаются клиническими проявлениями.
Гипофосфатемический нефролитиаз с остеопорозом. Выделяют два типа заболевания, оба наследуются аутосомно-доминантно и манифестируют во взрослом возрасте. Гипофосфатемический нефролитиаз с остеопорозом 1-го типа развивается вследствие мутаций в гене SLC34A1, 2-й тип заболевания обусловлен мутациями гена SLC9A3R1 (NHERF1). В гене SLC9A3R1 известна повторяющаяся в различных семьях мутация LEU110VAL (328C>G), остальные изменения генов SLC34A1 и SLC9A3R1 носят индивидуальный характер. В лабораторных показателях пациентов обращают на себя внимание гипофосфатемия, гиперфосфатурия, гиперкальциурия, увеличение уровня витамина D. Таким образом, мутации генов приводят к нарушению обмена фосфатов. Значительные потери фосфора обусловливают нарушение регуляции синтеза паратгормона и витамина D и сопровождаются гиперпаратиреозом, гиперкальциурией с формированием нефрокальциноза. Характерной особенностью течения заболевания является развитие у пациентов выраженного остеопороза, вплоть до состояния, характеризующегося частыми переломами и деформацией позвоночника. Это делает особенно актуальной превентивное ДНК-тестирование родственников, не имеющих каких-либо клинических проявлений заболевания, с целью своевременной коррекции у них минерального обмена и профилактики остеопороза [12].
Семейная гипомагниемия с гиперкальциурией и нефрокальцинозом – аутосомно-рецессивное заболевание, проявляющееся в раннем детстве (3–7 лет). Представлено двумя типами, принципиально отличающимися друг от друга наличием или отсутствием поражения глаз у пациентов. Мутации в гене CLDN16 (PCLN1) приводят к семейной гипомагниемии без поражения органа зрения, в то время как мутации гена CLDN19, напротив, сопровождаются зрительными проблемами.
Ген CLDN16 кодирует выработку белка клаудина 16, селективно экспрессируемого в плотных контактах клеток петли Генле (толстой части ее восходящего отдела), где играет ключевую роль в реабсорбции бивалентных катионов. В составе гена описано более 40 различных мутаций, наиболее частыми из которых являются Leu151Phe, обнаруженная у пациентов из Германии и Восточной Европы (эффект основателя), а также другая миссенс-мутация Ala139Val, характерная для жителей Северной Африки. Кроме того, мутация GLY198ASP (rs104893723) выявлена в двух неродственных семьях с заболеванием [13, 14].
В гене CLDN19 описано менее 10 мутаций, наиболее частая представляет собой миссенс-вариант Gly20Asp, обнаруженный у пациентов испанского и французского происхождения (предположительно эффект основателя).
Больные теряют с мочой массу магния и кальция, вследствие чего у них возникают вторичная гипомагниемия и нефрокальциноз. Уровень кальция в крови остается нормальным. Мутации генов CLDN16 и CLDN19 приводят к массивной потере магния почками по причине нарушения его транспорта [15]. Таким образом, диагноз базируется на триаде признаков, включающей гипомагниемию, гиперкальциурию и нефрокальциноз. У пациентов часто наблюдаются повторные инфекции мочевыводящей системы, полиурия, полидипсия и гематурия. В последующем у больных развиваются нефрокальциноз и почечная недостаточность. Показано, что у 33% пациентов с семейной гипомагниемией, обусловленной мутациями в гене CLDN16, к пубертату или в раннем взрослом возрасте развивается терминальная почечная недостаточность. При мутациях в гене CLDN19 заболевание протекает более агрессивно. Так, к пубертатному возрасту 66% пациентов с мутациями клаудина 19 находятся в терминальной стадии почечной недостаточности.
Зрительные нарушения при мутациях в гене CLDN19 включают миопию, пигментный ретинит, макулярную колобому, страбизм, астигматизм и нистагм.
Синдром Барттера представляет собой генетически гетерогенное заболевание, выделяют по меньшей мере пять типов болезни. Для 3-го (ген CLCNKB) и 4-го (гены CLCNKA,CLCNKB, BSND) типов синдрома Барттера нефрокальциноз не характерен. У пациентов с 1-м (ген SLC12A1), 2-м (ген KCNJ1) и 5-м (ген MAGED2) типами болезни развивается тяжелое состояние, проявляющее себя еще на уровне внутриутробного развития в виде увеличения количества околоплодных вод с последующим формированием нефрокальциноза у новорожденного ребенка. Первый и 2-й типы синдрома наследуются аутосомно-рецессивно, 5-й – Х-сцепленно-рецессивно. Клинические признаки заболевания проявляются сразу после рождения. Причиной синдрома Барттера считают нарушение функции почечных канальцев, проявляющееся снижением реабсорбции ионов Cl- (и соответственно, Na+) клетками восходящего отдела петли Генле. Патогенез синдрома Барттера до конца не выяснен.
К классическим признакам заболевания относят выраженные нарушения электролитного обмена (гипокалиемия), кислотно-щелочного равновесия (метаболический алкалоз), гиповолемию, компенсаторную гиперплазию юкстагломерулярного аппарата почек и вторичный гиперальдостеронизм [16]. Характерны низкий рост, полиурия, остеопения, парезы, судороги, нарушение умственного развития ребенка.
Дистальный канальцевый ацидоз. Первичная тубулопатия, наследуемая по аутосомно-доминантному типу, проявляет себя в раннем детстве (1–8 лет) и обусловлена мутацией в гене SLC4A1. При изучении дистального канальциевого ацидоза L. Bruce описал три мутации гена SLC4A1, затрагивающие один и тот же кодон гена: Аrg589Cys, Arg589His, Arg589Ser. Не исключено, что данный кодон служит так называемой горячей точкой гена SLC4A1. Остальные мутации в этом гене описаны как единичные случаи. Синдром проявляется метаболическим ацидозом, развивающимся вследствие нарушения подкисления мочи почками в отсутствие выраженного снижения функции клубочкового аппарата [17].
Стоит отметить, что мутации приведенного гена также могут способствовать развитию аутосомно-рецессивного дистального почечного канальциевого ацидоза с гемолитической анемией.
Кроме того, дополнительно выделяют некоторые другие аутосомно-рецессивные формы почечного канальциевого ацидоза, в частности сопровождающиеся нейросенсорным нарушением слуха (гены ATP6V0A4 и ATP6B1). Все перечисленные формы заболевания характеризуются формированием камней в почках, описаны случаи оперативного лечения данного типа МКБ в возрасте 12 и 20 лет.
Витамин D-зависимый рахит представляет собой генетически гетерогенное заболевание, выделяют два основных типа витамин D-зависимого рахита – I и II. Оба наследуются аутосомно-рецессивно и проявляют себя клинически в младенческом возрасте (0–2 года). I тип заболевания подразделяется на 2 варианта: Ia (обусловлен мутациями в гене CYP27B1 D3-1-альфа-гидроксилазы витамина D3) и Ib (связан с мутациями в гене CYP2R1 25-гидроксилазы витамина D) [18–20]. Оба гена участвуют в биохимическом преобразовании витамина D в его активную форму, а значит, имеют существенное значение для кальциево-фосфорного обмена в организме. То же касается и II типа заболевания, который также подразделяется на два подтипа – IIa и IIb. Первый подтип связан с мутациями в гене рецептора витамина D VDR, для второго ген пока не известен. Клиническая картина витамин D-зависимого рахита сводится к задержке физического и моторного развития, для больных характерны низкий рост, мышечная гипотония, снижение минеральной плотности костной ткани, костные деформации, переломы, боли в костях, нарушение роста волос вплоть до развития алопеции (у 75% больных), неврологическая симптоматика в виде судорог и повышенной возбудимости. У некоторых пациентов снижается слух [21]. В лабораторных показателях обращают на себя внимание гипокальциемия, гипофосфатемия, нормальный или повышенный уровень витамина D. Постепенно развивается вторичный гиперпаратиреоз, который формируется в ответ на длительное снижение уровня кальция в крови, проявляется избытком паратгормона, способствующего выведению из костной ткани кальция и фосфора. Со временем за счет массивного выхода кальция из костной ткани увеличивается выделение избытка кальция с мочой, что способствует камнеобразованию в почках (нефролитиаз) и отложению солей кальция в паренхиме почек (нефрокальциноз). Терапия заболевания сводится к длительному приему витамина D [22, 23].
Основная часть случаев заболевания связана с индивидуальными мутациями в семьях больных. Однако в гене VDR описаны повторяющиеся у неродственных больных мутации: ARG73GLN, TYR292TER, GLY33ASP, ARG77GLN, ARG47GLN, ARG30TER. Кроме того, в гене VDR выявлено несколько полиморфизмов, ассоциированных с развитием полигенного варианта МКБ (rs1544410 (BsmI), rs731236 (taq1), rs10735810 (rs2228570 – Fok1).
В гене CYP27B1 в 8-м экзоне также определена «горячая точка» 7-BP DUP CCCACCC. Данная мутация выявлена у больных витамин D-зависимым рахитом Iа-типа родом из Филиппин, Польши, Китая, США и Латинской Америки. Кроме того, у французских канадцев установлен эффект основателя для мутации 1-BP DEL, 958G гена CYP27B1 [24].
Также стоит упомянуть о существовании других форм рахита, которые могут сопровождаться камнеобразованием в почках. Например, наследственный гиперфосфатемический рахит с гиперкальциурией. Заболевание связано с мутациями в гене SLC34A3, кодирующем натрий-фосфатный котранспортер. От функции данного белка зависит поддержание концентрации неорганического фосфата на оптимальном уровне [25–27]. Патология характеризуется кодоминантным типом наследования и проявляется в первые годы жизни ребенка. Полный спектр симптомов развивается у обладателей двух мутаций гена в гомозиготной или компаунд-гетерозиготной форме. Показано, что гетерозиготные носители мутации гена SLC34A3 также имеют признаки заболевания, однако патология у них характеризуется более мягким течением. В крови таких носителей повышен уровень витамина D, в моче наблюдается гиперкальциурия. В отличие от пациентов с гомозиготной формой заболевания у гетерозиготных носителей мутации нет патологии со стороны скелета и характерных для рахита признаков. Гипофосфатемия при данном заболевании вторична и развивается на фоне значительной потери фосфата с мочой на уровне почек (страдает реабсорбция фосфата в почечных канальцах). В отличие от других типов гипофосфатемического рахита у пациентов с мутациями в гене SLC34A3 гиперкальциурия развивается на фоне повышенного содержания в крови витамина D и увеличения абсорбции кальция в кишечнике. Уровни кальция крови и паратгормона у больных, как правило, находятся в пределах референсных значений. Вследствие развития гиперкальциурии для больных характерно наличие кальциевого нефролитиаза [28, 29].
В семьях больных наследственным гипофосфатемическим рахитом с гиперкальциурией в основном описаны индивидуальные мутации. При выявлении мутаций гена SLC34A3 пациентам рекомендуется осуществлять динамический мониторинг содержания фосфата и витамина D в крови и моче, проводить терапию, направленную на поддержание минеральной плотности костной ткани, осуществлять профилактику осложнений остеопороза.
Идиопатическая абсорбционная гиперкальциурия. Еще одним интересным с точки зрения кальциевого обмена заболеванием является идиопатическая абсорбционная гиперкальциурия, наследуемая по аутосомно-доминантному типу. Примечательно, что заболевание может проявиться в виде уролитиаза в любом возрасте. Патология связана с мутациями в гене ADCY10 (SAC), кодирующем растворимую аденилатциклазу, задействованную в бикарбонатной регуляции c AMP опосредованного сигнального пути, необходимого для поддержания внутриклеточного pH и мембранного потенциала клетки [30]. Показано, что гиперкальциурия при мутациях гена ADCY10 ассоциирована с усиленным всасыванием кальция из поступающей в организм пищи на уровне кишечника [31]. Частых и повторяющихся мутаций в гене ADCY10 у больных не описано.
Для больных этой патологией характерно наличие оксалатно-кальциевых камней, а МКБ обычно имеет рецидивирующее течение [32].
Инфантильная гиперкальциемия 1-го типа. Кроме нарастания содержания кальция, связанного с увеличением его кишечной абсорбции, существуют и другие формы первичной гиперкальциемии. К таким заболеваниям относится инфантильная гиперкальциемия 1-го типа, связанная с мутациями в гене CYP24A1, кодирующем фермент 24-гидроксилазу, участвующую в катаболизме витамина D в мононуклеарах периферической крови [33]. Патология наследуется по аутосомно-рецессивному типу и характеризуется ранней манифестацией – обычно в первые месяцы жизни. Однако описаны случаи и более позднего возраста проявления заболевания (в 19 лет).
Мутации гена CYP24A1 снижают активность работы белка-продукта и в основном носят индивидуальный характер. К настоящему времени описана одна повторяющаяся мутация 427delGAA, обнаруженная у двух неродственных пациентов. В одном случае заболевание проявило себя нефрокальцинозом в младенчестве, во втором – первый эпизод камнеобразования в почках случился у пациента в 19 лет. При этом существует гипотеза, согласно которой ген CYP24A1 обладает не только вариабельной экспрессивностью, но и неполной пенетрантностью. Предполагается, что проявление генотипа в фенотипе зависит от количества поступающего в организм пациента витамина D и прием чрезмерных доз витамина D может потенцировать проявление заболевания. В случае наличия мутации, снижающей активность CYP24A1, деградация витамина D замедляется, что само по себе выступает причиной увеличения у таких пациентов уровня витамина D в крови, отсюда и гиперкальциемия. В связи с этим при выявлении мутации в гене CYP24A1 пациенту следует избегать употребления данного витамина [34].
Другие моногенные формы МКБ встречаются реже, и они менее изучены. Совершенно очевидно, что для диагностики моногенных форм уролитиаза недостаточно тестирования на наиболее частые мутации в определенных генах. Современные молекулярно-генетические технологии, такие как секвенирование, в частности NGS (nextgenerationsequencing), позволяют выявлять и редкие мутации, вызывающие камнеобразование. По всей видимости, на сегодняшний день именно они должны использоваться для выявления точной молекулярной причины заболевания. Стоит отметить, что с помощью молекулярно-генетических методов можно оценить риск развития уролитиаза не только для самого обследуемого, но и для его потомства. Таким образом, применение молекулярно-генетических методов при моногенной патологии позволяет идентифицировать весь спектр геномных нарушений, тем самым точно установить причину заболевания и предсказать ее появление в ряду поколений.
Вместе с тем даже при имеющейся возможности применения высокотехнологичных методов ДНК-тестирования диагностика МКБ может представлять определенные сложности. Это связано с поздним или неполным проявлением патологии у пациента, а также с наличием у больных других хронических заболеваний, маскирующих симптомы конкретного синдрома, что, несомненно, говорит о важности тщательного клинического обследования тестируемых.
В данной работе мы обобщили данные по исследованиям далеко не всех генов, влияющих на формирование мочевых камней. Несомненно, исследования по выявлению генных мутаций и новых генов-кандидатов в отношении развития МКБ продолжаются учеными разных стран ввиду актуальности МКБ. Развитие технологий анализа ДНК привело к разработке методов секвенирования нового поколения, что позволяет определять не только повторяющиеся и описанные ранее мутации, но и выявлять новые патогенные и вероятно патогенные варианты генов, вносящие значительный вклад в риск развития многих заболеваний человека, включая МКБ.
Основным препятствием к использованию этих технологий в клинической практике являлась их высокая стоимость для пациентов. Однако благодаря постоянно снижающейся стоимости, высокой производительности, а также широким возможностям мультиплексирования по образцам и тестируемым регионам технология NGS в ближайшем будущем должна прийти на смену целому спектру методов ДНК-диагностики. Трактовка полученных результатов таких исследований требует привлечения квалифицированных и опытных специалистов, разбирающихся как в генетических аспектах, так и в особенностях группы нарушений, для которой выполняется генетическое тестирование. В связи с этим совершенно очевидно, что для выяснения этиологических факторов в случае моногенных форм МКБ необходимо проведение медико-генетического консультирования. Выявить генетические нарушения, составить объективный прогноз и выбрать оптимальную схему лечения, а в ряде случаев и предотвратить заболевание – задача превентивной и персонализированной медицины будущего.


