
Важным элементом высококвалифицированной помощи в настоящее время все чаще становится медико-генетическое консультирование. Возможности современных молекулярно-генетических технологий позволяют разобраться в сложных диагностических ситуациях, установить этиологию заболевания, своевременно назначить лечение и обеспечить профилактику в семье больного. Молекулярно-генетические методы получили широкое распространение и в диагностике мочекаменной болезни (МКБ). Данное заболевание широко распространено как среди взрослого населения, так и среди детей. Распространенность нефролитиаза во взрослой популяции России составляет не менее 450 на 100 тыс. населения, у подростков – 80 на 100 тыс., у детей – около 20 случаев на 100 тыс. [1, 2]. В большинстве (80%) случаев у пациентов выявляются кальциевые конкременты. На цистиновый уролитиаз приходится 1–3% случаев МКБ [3].
Основную долю случаев МКБ можно отнести к многофакторной патологии, т.е. к болезням с наследственной предрасположенностью. Многофакторные заболевания возникают из-за полиморфизма генов, которые обусловливают генетическую предрасположенность к камнеобразованию в почках при воздействии факторов окружающей среды. Так, образование кальциевых конкрементов может быть связано с наличием полиморфизмов в генах кальциево-фосфорного обмена: VDR, CASR, CALCR, SLC34A1, CLDN14, TRPV5, TRPV6 и др. [4].
Меньшую долю составляют моногенные формы патологии. Как правило, это редкие заболевания, манифестирующие в раннем детском возрасте. Эта группа болезней связана с мутацией в одном гене. Несмотря на то что встречаются они редко, многие из них хорошо известны практикующим врачам-урологам. Ярким примером такого заболевания является цистинурия.
Цистинурия – это наследственное моногенное заболевание, которое характеризуется гиперэкскрецией цистина с мочой (более 400 мг/день при норме менее 30 мг/сут) [5–11]. Распространенность данного заболевания в мире составляет в среднем 1:7000 (от 1:2500 среди ливийских евреев до 1:100000 среди шведов) [12–14]. Первые клинические симптомы болезни появляются в возрасте от 2 до 40 лет [15]. Основным проявлением заболевания являются цистиновые камни, причем более чем в 75% случаев в почках образуются двусторонние камни [9]. Течение заболевания, как правило, рецидивирующее. Диагностика основывается на данных спектрального анализа мочевого камня и определения цистина в моче. Однако диагностика цистинурии у детей до 1 года представляет определенные трудности в связи с тем, что в этом возрасте система транспорта аминокислот в мембранах почечных канальцев незрелая и повышение уровня цистина в моче может быть транзиторным [16]. В связи с этим использование молекулярно-генетических методов приобретает особое значение, так как позволяет уточнить клинический диагноз и выявить молекулярную причину заболевания. Представляем собственное клиническое наблюдение.
Пациент М. 4 лет 7 месяцев поступил с жалобами на эпизоды фебрильной лихорадки и отхождение конкремента с мочой (со слов мамы). Ребенок рожден от 1-й беременности, роды срочные самопроизвольные с акушерским пособием (метод Кристеллера), масса тела при рождении – 4110 г, рост –56 см. Оценка по шкале Апгар – 8/9 баллов. Из анамнеза известно, что с 6 мес. у ребенка отмечалось повышение температуры тела до фебрильных цифр без катаральных явлений, выявлялась лейкоцитурия (до 32 клеток в поле зрения), проводилась антибактериальная терапия с положительным, но кратковременным эффектом. При УЗИ с 6 мес. выявлялся симптом «белых пирамидок» в обеих почках. При УЗИ яичек выявлены эхо-признаки двустороннего крипторхизма. В 9 мес. у пациента зафиксирован повторный эпизод лихорадки с лейкоцитурией. Выполнена цистография, по результатам которой диагностирован пузырно-мочеточниковый рефлюкс I–II степеней слева.
В биохимических анализах крови изменений уровня общего и ионизированного кальция, фосфора не наблюдалось. Однако при биохимическом исследовании суточной мочи определялась гиперкальциурия (Ca/Cr=0,44 мг/мг (норма до 0,21). При УЗИ выявлен медуллярный нефрокальциноз, размеры почек, толщина паренхимы, состояние кровотока без изменений. В 3 года у ребенка самостоятельно отошел почечный камень (гладкий желто-коричневого цвета). Результаты рентгенофазового анализа: оксалат кальция моногидрат (вевеллит) – 23%, оксалат кальция гидрат (ведделит) – 28%, дигидроксофосфат кальция (гидроксиапатит) – 32%, фосфат кальция (витлокит) – 17%.
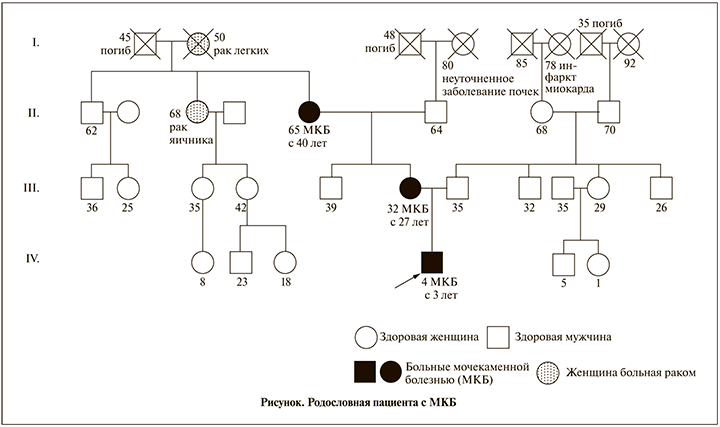
В семейном анамнезе пациента у ряда родственников выявлена МКБ. Так, у матери при УЗИ выявлены 3 камня размером до 3 мм в правой почке в возрасте 27 лет. У бабушки по материнской линии МКБ диагностирована в возрасте 40 лет (см. рисунок). При биохимическом анализе крови выявлен умеренный гипопаратиреоз (уровень паратиреоидного гормона составил 13,5 пг/мл при норме 16–62 пг/мл). Остальные показатели, отражающие фосфорно-кальциевый обмен, были в пределах референсных значений: Са общий максимально 2,66 мМоль/л (норма – 2,20–2,70), Са ионизированный – 1,26 мМоль/л (норма – 1,13–1,32), фосфор – 1,63 ммоль/л (норма – 1,3–2,26 ммоль/л), 25(ОН)D – 56,8 нг/мл (норма – 14–60), креатинин – 47 мкмоль/л. Расчетная скорость клубочковой фильтрации по формуле Шварца составила 98 мл/мин/1,73 м2.
β2-микроглобулин (как показатель низкомолекулярной протеинурии – дисфункции проксимального канальца) в моче не обнаружен.
Пациенту в возрасте 3,5 лет проведено секвенирование клинического экзома. В результате исследования в гене SLC7A9 выявлена мутация c.695A>G (p.Tyr232Cys, rs121908487) в гетерозиготной форме (генотип A/G, Tyr/Cys).
Ген SLC7A9 локализован в длинном плече 19-й хромосомы (19q13.1) и кодирует белок В(0,+)АТ, относящийся к семейству легких субъединиц аминокислотных транспортеров, работающих в почках, печени, тонкой кишке и плаценте [12, 13, 17–20]. Мутации гена SLC7A9 ответственны за развитие цистинурии типа В. У пациента выявлена ранее описанная мутация c.695A>G, которая располагается в 6-м экзоне гена SLC7A9. Данная однонуклеотидная замена приводит к замене тирозина на цистеин в 232-м положении молекулы белка – продукта гена. Частота данной мутации в популяции Европы составляет 0,02%. Обычно данная мутация в сочетании с другим патогенным вариантом того же гена приводит к клинической манифестации заболевания в виде образования цистиновых камней.
Ввиду того, что у больного были выявлены кальциево-оксалатные камни, принято решение провести дополнительное молекулярно-генетическое исследование (см. таблицу).

В 8 из 15 проанализированных генов выявлены полиморфизмы, ассоциированные с нарушением кальциево-фосфорного обмена. Таким образом, клиническая картина, обусловленная наличием кальциевых конкрементов, может быть связана с генетическими особенностями пациента.
В представленном клиническом наблюдении при уточнении молекулярной причины заболевания возникли трудности, связанные с несоответствием результатов первоначального молекулярно-генетического анализа (исследование клинического экзома) и клинических проявлений МКБ у пациента.
Так как у пациента был диагностирован кальциево-оксалатный нефролитиаз, первоначально назначено исследование для подтверждения моногенной формы нарушения кальциево-фосфорного обмена. Однако результаты секвенирования клинического экзома не выявили клинически значимых мутаций, отвечающих за нарушение кальциевого обмена. Вместе с тем у пациента выявлено гетерозиготное носительство мутации в гене SLC7A9, отвечающем за аутосомно-рецессивную форму цистинурии типа В.
Мутация c.695A>G (p.Tyr232Cys, rs121908487), обнаруженная у пациента, относится к так называемым мутациям type non-I, которые в гетерозиготном состоянии обычно не приводят к образованию цистиновых камней [21]. Возможно, у таких пациентов (гетерозиготных носителей) имеется нерегистрируемое в ходе биохимических исследований увеличение уровня цистина в моче. Как известно, определить наличие цистина в моче весьма сложно из-за выраженной нестойкости цистина при контакте с окружающей средой. Однако есть данные, согласно которым мутации в гене SLC7A9 в гетерозиготном состоянии могут способствовать образованию камней преимущественно кальциево-оксалатного типа. Это связано с тем, что цистин мочи сам по себе способствует кристаллизации кальция в мочевыводящих путях [22].
Кроме того, образование кальциево-оксалатных камней может быть обусловлено полиморфизмом генов, отвечающих за кальциево-фосфорный обмен. Поэтому пациенту было проведено генетическое исследование по панели генов, ассоциированных с образованием кальциевых камней. Данное исследование показало наличие у больного полиморфизмов, дополнительно способствующих образованию кальциево-оксалатных конкрементов.
Уточнение молекулярно-генетической картины заболевания дало возможность разработать схему персонализированного лечения и обеспечить профилактику МКБ в семье больного. Прежде всего это неспецифические, но крайне важные рекомендации по увеличению количества потребляемой воды (около 2 л/1,73 м2, в том числе около ¼ в ночное время) и соблюдению низкосолевой диеты (для ребенка не более 2–3 г/сут, для взрослых не более 5 г/сут пищевой соли, в т.ч. содержащейся в готовой еде – такой, как хлеб, мясопродукты, сыр и т.п). Кроме того, рекомендовано ограничение метионин-содержащих продуктов (сыр, яйца, рыба) в связи с метаболизмом метионина в цистеин. Необходим также курсовой прием алкалинизирующих смесей, в том числе как донация цитрата, важнейшего ингибитора кристаллообразования, под контролем рН мочи (целевой уровень не выше 7–7,2 с учетом многофакторной причины камнеобразования). Лечение препаратами, связывающими цистин (тиопронин, D-пеницилламин), не было назначено в связи с недоступностью в РФ количественного определения цистина в порциях мочи, а также из-за возможных побочных эффектов препаратов. Кроме того, учитывая полиморфизм генов, ответственных за кальциево-фосфорный обмен, и риск развития гиперкальциемии, не назначался гипотиазид для снижения степени кальциурии [23, 24].
Мы представили сложное клиническое наблюдение ребенка 4 лет, страдавшего МКБ, у которого причиной заболевания стало сочетание носительства мутации в гене SLC7A9, ответственном за цистинурию типа В, и генетической предрасположенности к образованию кальциевых конкрементов. Выявленные генетические особенности позволили определиться с тактикой ведения пациента и сформировать подход к проведению профилактики камнеобразования в семье пациента.


